武汉大学刘胜院士领导的团队---β-Ga₂O₃ 界面上的金属接触和肖特基势垒: 高通量辅助第一性原理计算
武汉大学刘胜院士、郭宇铮教授和张召富教授团队在学术期刊 Journal of Applied Physics 发布了一篇名为 Metal contacts and Schottky barriers at β-Ga2O3 interfaces: High-throughput-assisted first-principles calculations(β-Ga2O3 界面的金属接触和肖特基势垒:高通量辅助第一性原理计算)的文章,该篇内容被 Journal of Applied Physics 评选为编辑推荐(Editor’s pick)文章。
1. 项目支持
该项研究得到了国家自然科学基金(NNSFC)(Grant Nos. 62174122、52302046、U2241244、62361166628 和 L2424216)、广东省基础与应用基础研究基金(Grant Nos. 2024A1515011764、2022A1515110149 和 2024A1515010383)、武汉曙光知识创新计划(Grant No. 2023010201020262)、江苏省自然科学基金(Grant No. BK20230268)以及湖北省电子制造与封装集成重点实验室(武汉大学)开放基金(Grant No. EMPI2024020)的支持。文中的计算工作由武汉大学超算中心提供支持。
2. 背景
β-Ga2O3 作为超宽带隙半导体(带隙~4.8 eV),凭借高击穿电场(8 MV/cm)和低成本制备等优势,成为新一代功率器件的理想候选材料。然而,β-Ga2O3 器件性能受限于金属/β-Ga2O3 界面特性,尤其是肖特基势垒高度(SBH)。β-Ga2O3 的晶体结构复杂,含不同配位的 Ga/O 原子,导致其具有各向异性,且由于费米能级钉扎效应的影响,使得传统 Schottky-Mott 模型难以适用。而实验得到的金属/β-Ga2O3 界面的 SBH 与金属功函数的关系存在矛盾,且理论研究多聚焦于 β-Ga2O3 的单一晶面或少数金属。亟需系统性研究 β-Ga2O3 不同晶面与金属组合的界面特性,以指导 β-Ga2O3 基器件中电极材料的设计。
3. 文章摘要
金属电极与 β-Ga2O3 形成的界面是 β-Ga2O3 基电子和光电器件的关键组成部分。虽然已有少数关于金属/β-Ga2O3 界面电学性质的研究,但这些研究主要聚焦于单一晶面的 β-Ga2O3 或少数几种金属。本研究通过高通量界面预测与生成方案,从数千万种候选组合中自动筛选出具有最小晶格失配率和界面面积的九种金属/β-Ga2O3 界面体系,并基于第一性原理计算系统研究了这些界面的金属接触特性。计算结果表明,金属/β-Ga2O3 界面的肖特基势垒高度(SBHs)与现有实验结果具有良好的一致性。其中,Al/β-Ga2O3 (100)、Ti/β-Ga2O3 (100)、Ni/β-Ga2O3 (100) 和 Co/β-Ga2O3 (-201) 界面表现出较低的n型肖特基势垒和较高的电子转移效率,表明 Al、Ti、Ni 和 Co 可作为理想的欧姆接触电极材料。更重要的是,还获得了多个具有优异接触特性的金属/β-Ga2O3 界面原子结构,这些结构在理论和实验研究中均未被报道。这些发现为 β-Ga2O3 功率器件中金属电极材料的理性选择及器件性能优化奠定了理论基础。
4. 计算细节
•本研究所有计算均基于密度泛函理论(DFT)。具体而言,肖特基势垒高度(SBHs)的计算使用 QuantumATK 软件完成,其他计算任务通过 VASP 软件包实现。
•采用 Perdew-Burke-Ernzerhof(PBE) 交换关联泛函结合投影缀加波赝势,对金属/β-Ga2O3 界面原子结构进行优化,直至体系能量和原子受力分别收敛至 1.0×10-5 eV 和 0.01 eV/Å 阈值。
•鉴于 PBE 方法对 β-Ga2O3 带隙存在显著低估问题,金属/β-Ga2O3 界面电学性质的计算采用 Heyd-Scuseria-Ernzerhof(HSE)杂化泛函以获得更精确的结果。
5. 创新点
•获得了多个具有优异接触特性的金属/β-Ga2O3 界面原子结构,这些结构在理论和实验研究中均未被报道。
•发现 Al/β-Ga2O3 (100)、Ti/β-Ga2O3 (100)、Ni/β-Ga2O3 (100) 和 Co/β-Ga2O3 (201) 具有相对较低的 n 型肖特基势垒和较高的电子转移效率,可作为欧姆接触电极。
6. 结论
研究团队采用高通量 IPG 方案构建并系统研究了九种具有最小晶格失配率和界面面积的金属/β-Ga2O3 界面接触特性。结果表明,由于 β-Ga2O3(-201)晶面的不对称性,Sc/β-Ga2O3 和 Co/β-Ga2O3 界面表现出最小的层间距和最低的结合能。不同金属形成的 β-Ga2O3 界面肖特基势垒高度(SBHs)相似,表明金属/β-Ga2O3 界面存在强钉扎效应。此外,n 型 SBHs 与金属功函数呈现显著线性关系(钉扎因子0.17),与现有实验结果及经验值高度吻合,表明基于 IPG 方法构建的金属/β-Ga2O3 界面原子结构能够有效捕捉强钉扎效应。理论计算进一步揭示了费米能级钉扎源于 β-Ga2O3表面态与金属诱导带隙态的共同作用。值得注意的是,Al/β-Ga2O3 (100)、Ti/β-Ga2O3 (100)、Ni/β-Ga2O3 (100) 和 Co/β-Ga2O3 (-201) 界面具有较低的n型肖特基势垒和较高的电子转移效率,表明Al、Ti、Ni 和 Co 作为欧姆接触电极的潜力。本研究不仅为 β-Ga2O3 功率器件金属电极的理性选择提供了重要理论依据,同时验证了 IPG 方案在高质量半导体界面设计中的普适性和有效性。
7. 图文内容
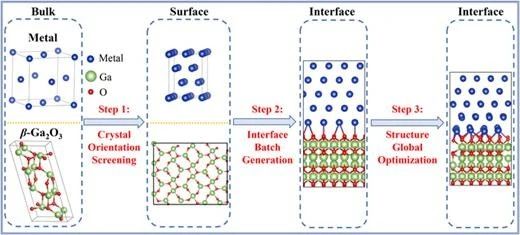
图 1. 金属/β-Ga2O3 界面结构高通量界面预测与生成方案的工作流程。
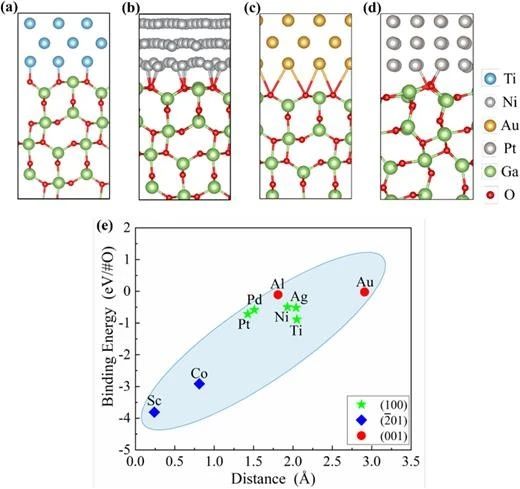
图 2. (a) Ti (100)/β-Ga2O3 (100)、(b) Ni (100)/β-Ga2O3 (100)、(c) Au (100)/β-Ga2O3 (100) 和 (d) Pt (100)/β-Ga2O3 (001) 界面的原子结构。(e) 界面结合能与层间距离的关系。
DOI:
doi.org/10.1063/5.0256577
本文转发自《亚洲氧化镓联盟》订阅号